
9 岁女孩转氨酶异常 2 年查不出病因!竟是体内铜 “困住” 排不出,最终确诊罕见的肝豆状核变性
发布时间:2025/6/19 15:29:44 / 【关闭】
在人体众多微量元素中
铜,对人体健康至关重要
它不仅是多种重要酶的组成部分
还参与能量代谢、铁代谢、
神经系统发育等重要生理过程
然而,当它在体内出现代谢异常
竟能引发致命疾病
今天,我们将通过
青岛市妇女儿童医院儿童肝病门诊
救治的一例铜代谢障碍性疾病 ——
肝豆状核变性患儿的故事
揭开这种罕见病的神秘面纱

01 肝功异常 2 年病因未明
另一指标显著异常引起了医生警惕
9岁患儿彤彤(化名)病情颇为曲折:2 年前因肺炎在当地医院首次发现谷丙转氨酶(ALT)高达 276U/L(正常参考值 < 40U/L),虽规律口服保肝药物治疗 1 年余,但后续多次复查肝功能始终未恢复正常。
由于彤彤除肝酶升高外,无黄疸、神经系统症状等典型表现,病因长期未明。
来青前一周,当地医院复查提示肝酶再次异常,家属经多方咨询后转至青岛市妇女儿童院儿童肝病门诊。
入院后关键检查显示:铜蓝蛋白检测值 < 0.023g/L(正常值 0.2-0.6g/L),这一指标的显著异常引起了救治团队的警惕!
02 系统检查锁定罕见病因
潜伏两年的疑难病例被揪出
感染科儿童肝病团队以 "铜代谢异常" 为核心线索,展开系统性筛查诊断:
代谢指标深度筛查:24 小时尿铜检测结果突破儿童诊断阈值(>40μg/24h),证实铜排泄异常;
多学科联动排查:头颅 MRI 早期识别潜在神经系统受累,眼科裂隙灯检查明确角膜 K-F 环(铜沉积特征性表现);
基因诊断一锤定音:ATP7B 基因检测发现复合杂合突变,完全符合肝豆状核变性(Wilson 病)的遗传学特征。
最终,这例潜伏两年的疑难病例被确诊为常染色体隐性遗传的铜代谢障碍性疾病 —— 肝豆状核变性。
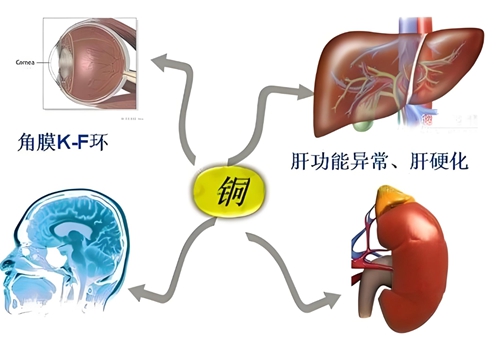
△图片来源于网络
03 个体化治疗打破 "铜枷锁"
需长期持续随访
结合患儿病情,儿童肝病门诊团队制定了精准的治疗方案:
驱铜治疗+饮食指导:采用药物促进铜排泄,制定低铜饮食方案;
全周期管理体系:建立定期复查机制,动态监测肝功能、铜代谢指标及并发症。
经规范治疗后,患儿肝酶指标已全面恢复正常,目前处于持续随访中。
疾病危害大,易被误诊
肝豆状核变性是由于 ATP7B 基因缺陷导致铜转运蛋白功能异常,引发铜在肝脏、脑、角膜等器官蓄积的罕见病。其临床特点包括:
起病隐匿多变:儿童期常见无症状肝酶升高,可进展为肝硬化或急性肝衰竭;
神经系统受累:部分患儿后期出现震颤、肌张力异常等神经症状;
早期漏诊率高:因缺乏典型症状,易被误诊为普通肝炎或保肝治疗无效的 "不明原因肝损伤"。
健康提醒
肝豆状核变性作为一种铜代谢障碍性疾病,因铜在体内异常蓄积致病,目前临床建议患者每日饮食铜摄入量控制在 1~1.5mg。
青岛市妇女儿童医院儿童肝病门诊专家提醒:
家长需监督肝豆状核变性患儿严格执行低铜饮食管理:避免食用动物内脏、贝类海鲜、坚果类(如花生、核桃)、豆类制品(如黄豆、豆腐)、菌菇类(如香菇、平菇)及巧克力等高铜食物,从饮食源头减少铜摄入;
同时需遵医嘱规律服用驱铜药物,确保治疗的连贯性。
由于肝豆状核变性具有家族遗传倾向,患者亲属存在较高患病风险。若家中儿童青少年确诊该病,需尽快对家庭成员开展筛查,以实现早期诊断与干预。
此外,对于有肝病家族史或出现不明原因肝功能持续异常的儿童,应尽早到专科门诊就诊。
青岛市妇女儿童医院已建立 “儿童不明原因肝酶升高追踪机制”,对持续异常病例常规开展铜代谢筛查及罕见病鉴别诊断,目前已成功诊治希特林蛋白缺乏症、Gilbert 综合征、Shwachman-Diamond 综合征等多种罕见肝病。该院通过 “早发现、精诊断、优管理” 的诊疗模式,为儿童肝脏健康构建起精准防护体系。